Federal Inspections and Enforcement
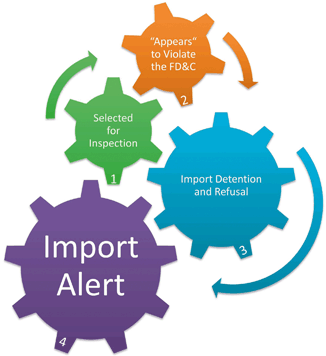
Fig. 2.1
Three marks of inspection utilized by the USDA/FSIS
The processing plants experienced a similar inspection. Processing initially involved cutting and boning whole carcasses along with the production of meat products like ham or bacon. These functions were usually completed in a facility adjacent to the slaughtering facility. The focus for FSIS in processing was on the overall production line, not the individual products. The emphasis was on sanitary conditions, which would contaminate meat previously inspected and approved.
For nearly 90 years, the FSIS inspected for disease using organoleptic methods . As with the original passage of the 1906 Act only crisis compelled changes to how the USDA conducted inspections. The 1990 E. Coli O157:H7 outbreak linked to the fast-food chain “Jack In the Box ” brought a new inspection concept to FSIS. Prior to the outbreak FSIS explored ways to modernize its inspection system. The surge in establishments, the increasing range of products, and the emergence of new technologies, ingredients, and processes proved too complex for FSIS. FSIS simply was overwhelmed and the 1990 outbreak highlighted the extent of the gaps in its surveillance. In response, FSIS underwent structural changes and developed a new rule for inspectors known as “Pathogen Reduction/Hazard Analysis and Critical Control Point System” (HACCP) . More than two decades later, HACCP remains the industry standard for FSIS inspections.
2.2.4 Organization and Evolution of FSIS
FSIS did not start out with a clear name or mission. As discussed in Chap. 1 and shown in Fig. 1.7, FSIS began as the Bureau of Animal Industries . Inspection functions were housed in this subagency from 1906 to 1953. President Eisenhower kicked off a lengthy series of changes; first, moving inspection functions to the Agricultural Research Service (ARS) . In 1968, when poultry inspections were added, the subagency was named Consumer and Marketing Services , a subdivision within the ARS. In a short two-year-span, inspections were first moved to the Animal & Plant Health Service in 1971, renamed APHIS in 1972, and then moved to a new subagency in 1982 called Food Safety & Quality Service . The final move came in 1981 when FS&QS was reorganized into FSIS (see Fig. 2.2). A great deal of upheaval for any organization, the subagency experienced over five large organizational shuffles in less than 30 years.

Fig. 2.2
Evolution of FSIS and unification of USDA inspections
Although FSIS’s exercises a narrower scope of authority, it utilizes a complex organizational structure. Visiting the FSIS’s website and explaining its organization, one can become easily lost. The main office to focus on is the Office of Field Operations (OFO) and Office of Investigation, Enforcement and Audit (OIEA) . The OFO manages all FSIS inspections and initiates the corresponding enforcement actions. FSIS deploys approximately 8000 FSIS inspectors and staff to about 6200 meat slaughtering and/or processing plants nationwide. The OFO, like the FDA’s ORA , organizes its inspectors into districts (see, Sect. 3.2 below). FSIS operates ten districts (see Fig. 2.3). Each district is overseen by a district manager (DM) and deputy district manager (DDM) . Both would be involved in serious enforcement actions.
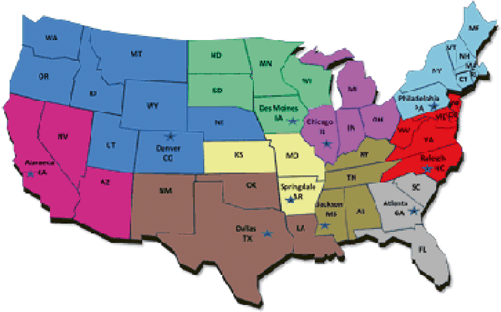
Fig. 2.3
US map broken into the ten OFO regions
The OIEA supports the OFO by conducting both criminal violations and investigating outbreaks. While OFO is focused on in-plant activities OIEA casts its attention toward in-commerce products. In particular, it investigates criminal violations and instances of intentional contamination. It will also play a key role in investigating foodborne illness outbreaks. Since OFO is limited to inspecting domestic facilities OIEA also verifies imported meat, poultry, and egg products meet applicable standards. This is a small sampling of the primary activities charged to the OIEA.
2.2.5 Enforcement Toolkit
Overview of Types of FSIS Enforcement Actions
FSIS enforcement options can be divided into three groups. The three groups or classes of enforcement actions as defined in the regulations are: regulatory control action , withholding action , and suspension . Each is defined in 9 CFR 500.1 as provided below. As can be seen in the excerpt of 500.1 below the enforcement actions escalate in severity. There is a final enforcement action, which is irreversible and in many ways the culmination of FSIS enforcement. That is the Withdrawal of Inspection under 9 CFR 500.6. Once inspectors are withdrawn from a facility, the facility cannot operate or re-open.
9 CFR 500.1
a.
A “regulatory control action” is the retention of product, rejection of equipment or facilities, slowing or stopping of lines, or refusal to allow the processing of specifically identified product.
b.
A “withholding action ” is the refusal to allow the marks of inspection to be applied to products. A withholding action may affect all product in the establishment or product produced by a particular process.
c.
A “suspension ” is an interruption in the assignment of program employees to all or part of an establishment.
Regulatory Control Actions
Regulatory controls actions are the mostcommonly used by FSIS inspectors. Regulatory control actions function as a low-level enforcement action that allows inspectors to correct an issue before a product leaves the facility or an equipment is reused. The violations are minor and the enforcement action is taken immediately. There are four scenarios provided in 9 CFR 500.2 when a regulatory control action may be taken. The four scenarios are provided below.
9 CFR 500.2(a)(1)-(4)
1.
Insanitary conditions or practices;
2.
Product adulteration or misbranding;
3.
Conditions that preclude FSIS from determining that product is not adulterated or misbranded; or
4.
Inhumane handling or slaughtering of livestock.
The focus for regulatory control actions centers on preventing noncompliant products from leaving the facility. This includes potential contamination or adulteration as well as misbranding. The USDA FSIS Rules of Practice Regulation (RPR) provides examples of each of the four conditions (Rules of Practice).2 The first three focus on ensuring that the products are safe and wholesome and the facility’s compliance program fully functioning. For example, ensuring equipment is clean (500.2(a)(1)), water does not collect on or around meat (500.2(a)(2), or the facility is well lit in order to allow inspectors to assess products and processes (500.2(a)(3)).
Regulatory control actions only result in a temporary delay. Typically product is retained and potentially reinspected. In other cases, equipment or facilities are closed until cleaned or repaired. In most instances, slaughter or processing lines are slowed or stopped temporarily.
The final basis for a regulatory control action finds its roots in a second enabling act. Congress originally passed the Humane Methods of Slaughter Act in 1958 (7 U.S.C. 1901 et. sEq. )3 The HMSA was updated in 1978 and provided the USDA FSIS authority to stop a slaughtering line until the abuses were corrected. The HMSA and 500.2(a)(4) do not apply to the slaughter of chickens or other poultry, only to livestock such as sheep, pigs, or cattle. The USDA/FSIS have issued a number of regulations , directives, and guidance to industry on how to humanely slaughter and handle livestock (9 C.F.R 313; FSIS Compliance Guide).4 A word of caution, if a reader is new to FSIS inspections, then be aware of enforcement reports and regulations in this area, in particular on inhumane handling and slaughter, can often be unsettling and graphic.
Although regulatory control actions are immediate, the facility still must be notified. The RPR makes clear the notification , typically via a Noncompliance Record (NR) , may be provided to the facility after the action is taken. This allows the hazard to be contained and the facility notified of a potential gap in its compliance program . In some cases, the facility may seek an appeal of the enforcement action. This appeal is taken to the next level of FSIS supervision.
Withholding Action or Suspension Without Notification
The remaining two categories of enforcement action can occur under two scenarios. Withholding actions refer to withholding the marks of inspection, which every product requires to enter the market legally. Suspension of inspection activities as the name suggests involves suspending inspectors and effectively stopping all production. Suspension differs from the most severe enforcement action—Withdrawal of Inspection . The Withdrwal of Inspection terminates FSIS inspections permanently and shutters the facility. Suspensions and withholding actions are similar, but a suspension will be in effect for longer than a withholding action.
There are certain violations FSIS deems as requiring enforcement action in these two categories immediately and without any prior notification to the facility. Subsection 500.3 provides four triggers for a withholding or suspension action without prior notice . Those are provided below.
9 CFR 500.3(a)(1)-(4)
a.
FSIS may take a withholding action or impose a suspension without providing the establishment prior notification because:
1.
The establishment produced and shipped adulterated or misbranded product as defined in 21 U.S.C. 453 or 21 U.S.C. 601;
2.
The establishment does not have a HACCP plan as specified in Sec. 417.2 of this chapter;
3.
The establishment does not have Sanitation Standard Operating Procedures as specified in Secs. 416.11–416.12 of this chapter;
4.
Sanitary conditions are such that products in the establishment are or would be rendered adulterated;
5.
The establishment violated the terms of a regulatory control action;
6.
An establishment operator, officer, employee, or agent assaulted, threatened to assault, intimidated, or interfered with an FSIS employee; or
7.
The establishment did not destroy a condemned meat or poultry carcass, or part or product thereof, in accordance with part 314 or part 381, subpart L, of this chapter within 3 days of notification .
The most common basis for withholding or suspension actions involves a serious and imminent threat to public health. Protecting the public health provides the primary rationale for taking a significant enforcement step without notification . In the RPR, FSIS directs inspectors to document the imminent threat when taking action under 500.3(a). Inspectors are also required to notify the facility orally and in writing “as promptly as the circumstances permit…” (Rules of Practice).5
The decision to take withholding and suspension actions come from higherspacelevels of authority. The decision to take a withholding action originates with inspectors in the plant, but must be made by the inspector in charge (ICC) or the frontline supervisor . In some cases, the decision is made by the district office . Suspension decisions on the other hand may only be made by the district office.
There are other grounds for taking enforcement action without prior notification that lack an urgency to protect public health. For instance, if any regulatory control action is not corrected or is repeated, then FSIS may take a withholding or suspension action without notification. In a sense, the facility already received notification through the regulatory control action and the regulations .
It is important to highlight instances where notification may be withheld that do not directly relate to food safety. Namely, the ability to withhold notification where FSIS personnel are confronted and possibly assaulted. Needless to say, it can be a contentious environment operating a facility with constant regulatory supervision. FSIS relies on the ability to work continually and freely in a facility. If an environment is created where inspectors do not feel comfortable to perform their duties, then enforcement action without notification works to restore the trust between FSIS and the host facility.
Withholding Action or Suspension with Prior Notification
If there is no immediate threat to public health then withholding or suspension actions require notification . Subsection 500.4 provides the criteria for withholding or suspension actions that require notification. Those are provided below. Prior to withholding the marks FSIS must provide written notice it intends to either withhold the marks of inspection or suspend inspections.
9 CFR 500.4(a)-(e)
FSIS may take a withholding action or impose a suspension after an establishment is provided prior notification and the opportunity to demonstrate or achieve compliance because:
a.
The HACCP system is inadequate, as specified in § 417.6 of this chapter, due to multiple or recurring noncompliances;
b.
The Sanitation Standard Operating Procedures have not been properly implemented or maintained as specified in §§ 416.13 through 416.16 of this chapter;
c.
The establishment has not maintained sanitary conditions as prescribed in §§ 416.2–416.8 of this chapter due to multiple or recurring noncompliances;
d.
The establishment did not collect and analyze samples for Escherichia coli Biotype I and record results in accordance with § 310.25(a) or § 381.94(a) of this chapter;
e.
The establishment did not meet the Salmonella performance standard requirements prescribed in § 310.25(b) or § 381.94(b) of this chapter.
Enforcement actions taken under 500.4 involve notification largely because it involves repeated noncompliance. Unlike 500.3 where there is no HACCP or standard operating procedures (SOPs) , 500.4 involve deficiencies in the compliance program . If these were one-off errors in the program, then they would most likely be caught in a regulatory control action . Section 500.4 instead aims for the gaps in the compliance program that result from inadequate procedures or processes. As such, the RPR directs inspectors to compile “extensive information” to provide both a factual basis for the facility to analyze and challenge and to demonstrate a pattern or history of failed corrective or preventative actions (Rules of Practice).6 Once presented with the notification and supporting evidence a facility is given an opportunity to respond by identifying areas of disagreement or share an interpretation of the regulations . This is in many ways similar to the Form 483 used by the FDA, which will be discussed in the Sect. 3.4 below.
Withdrawal of Inspection
Withdrawal of FSIS inspectors represents the pinnacle of the agency’s enforcement powers. There are several bases for withdrawing inspectors, which includes all of the previous actions that lead to withholding or suspension actions.
9 CFR 500.6
The FSIS Administrator may file a complaint to withdraw a grant of Federal inspection in accordance with the Uniform Rules of Practice, 7 CFR subtitle A, part 1, subpart H because:
a.
An establishment produced and shipped adulterated product;
b.
An establishment did not have or maintain a HACCP plan in accordance with part 417 of this chapter;
c.
An establishment did not have or maintain Sanitation Standard Operating Procedures in accordance with part 416 of this chapter;
d.
An establishment did not maintain sanitary conditions;
e.
An establishment did not collect and analyze samples for Escherichia coli Biotype I and record results as prescribed in § 310.25(a) or § 381.94(a) of this chapter;
f.
An establishment did not comply with the Salmonella performance standard requirements as prescribed in §§ 310.25(b) and 381.94(b) of this chapter;
g.
An establishment did not slaughter or handle livestock humanely;
h.
An establishment operator, officer, employee, or agent assaulted, threatened to assault, intimidated, or interfered with an FSIS program employee; or
i.
A recipient of inspection or anyone responsibly connected to the recipient is unfit to engage in any business requiring inspection as specified in section 401 of the FMIA or section 18(a) of the PPIA.
The slaughter and processing of meat is uniquely viewed as a privilege not a right. Unlike other food facilities, FSIS regulated facilities must apply for a grant of inspection . Think of it as applying for a driver’s license. And like a driver’s license can be revoked, so can a grant of inspection. Abuse the privilege, lose the privilege. The process to take revoke the grant of inspection can be lengthy. It not only involves a documented history of noncompliance, but a hearing before an Administrative Law Judge (ALJ) . A hearing preserves due process (see, Sect. 2.2.6 below). This process, and truly the entire grant of inspection feature, is unique to FSIS. The FDA while requiring a facility register prior to beginning operations is unable to bar a facility from beginning operations like FSIS can. This level of control requires careful checks to ensure the privilege is properly revoked.
2.2.6 Lessons from FSIS’s History— Food Law Is a Floor Not a Ceiling
There are a number of lessons to take away from the history of FSIS. In particular, note the slow pace of change in the inspection methods. Despite rapid changes in technology, demand, and the range of products, FSIS clung to outdated inspection criteria. There were rumblings of change prior to adopting HACCP , but it still took crisis to create change. If compliance is seen as a floor, then this can lead a facility into a false sense of security. As facilities struggle with the balancing marketability and compliance, it is important to look at the headlines. No facility wants to be associated with the crisis that leads to new rules or regulations .
Case Study: New Poultry Inspections Rule; First Change in Over 50 Years
An excellent example of the pace of change comes in a recent rule change announced by the USDA/FSIS. In the summer of 2014, the USDA/FSIS announced a new rule to poultry inspection. The new rule replaced the inspection model used when the PPI was adopted in 1957. For nearly 60 years, FSIS did not require facilities to test for Salmonella and Campylobacter . Under the new rule, known as the New Poultry Inspection System (NPIS) , facilities will be required to take preventative measure against Salmonella and Campylobacter contamination (NPIS Final Rule).7 This will include mandatory testing at two points in the production process. The NPIS will leave unchanged the maximum line speeds, which are currently 140 birds per minute. Although the speed sounds dizzying, pilot programs set maximum line speeds of 175 birds per minute.
Startling to consider how much the scientific knowledge of these two pathogens changed over 60 years or to think how the industry developed in that timeframe. Yet rather than elect for incremental changes the current model of regulation waits and issues sweeping regulations in an attempt to catch-up.
The history of FSIS also provides insight into political priorities. As governmental agencies, the USDA and FDA are only as effective as properly funded. Shifting an agency around, renaming and altering responsibilities does not speak to a well-regarded agency. The numbers support the notion. Not only are meat and poultry inspections deemed suitable for shuffling, but also for basic funding. Numerous studies of budget appropriations conclude the meat inspection budget either remains stagnant or contracts even in the face of a swelling mandate. Simply, consumers are demanding more meat and poultry, but the budget appropriations to ensure the safety of that meat and poultry is not gaining approval.
If the goal of an agency is consumer confidence than the agency name matters. When one hears the name Food Safety and Inspection Service a clear mandate emerges without knowing anything else about the agency. A name like Agricultural Research Service or Animal and Plant Health Service signals little about what the agency does. Would one really expect an agency named Agricultural Research Service to ensure the safety of meat products? The US Federal government is vast and names matter. Names provide clarity to the public about an agency’s mission and primary functions.
2.3 Overview of FDA Inspection Process and Enforcement Tools
2.3.1 Evolution of Inspection Authority
The FDA conducts warrantless inspections of the premises of regulated industries. The inspections may be “for cause” such as an inspection during a recall or adverse event or simply “surveillance” inspections as required by the Act. In either case, the FDA inspectors arrive unannounced and request total access to a facility for a period of four or more days. The FDA did not always enjoy the authority to inspect facilities. The 1906 Act, at only five pages, made no explicit reference to an ability to inspect facilities. An agent could arrive at a facility and request entry. If refused the FDA would need to go to court and obtain a warrant . With the 1938 Act Congress worked to patch-up this oversight. The 1938 Act created Section 704 to authorize the FDA to inspect facilities and added refusal to consent to inspect to the list of prohibited acts in Section 331 . As a prohibited Act, a refusal became a crime, typically a misdemeanor (see, Chap. 7).
The Supreme Court struck down the penalty in the 1938 Act. The court determined the provision in Section 704 which allowed consent, but penalized for withdrawing consent, as too vague to be enforceable.
UNITED STATES v. CARDIFF, 344 U.S. 174 (1952)
MR. JUSTICE DOUGLAS delivered the opinion of the Court.
Respondent was convicted of violating 301 (f) of the Federal Food, Drug, and Cosmetic Act, 52 Stat. 1040, 21 U.S.C. 331 (f). That section prohibits “The refusal to permit entry or inspection as authorized by section 704.” Section 704 authorizes the federal officers or employees “after first making request and obtaining permission…of the owner, operator, or custodian” of the plant or factory “to enter” and “to inspect” the establishment, equipment, materials and the like “at reasonable times.”
Respondent is president of a corporation which processes apples at Yakima, Washington, for shipment in interstate commerce. Authorized agents applied to respondent for permission to enter and inspect his factory at reasonable hours. He refused permission, and it was that refusal which was the basis of the information filed against him and under which he was convicted and fined… The Court of Appeals reversed, holding that 301 (f), when read with 704, prohibits a refusal to permit entry and inspection only if such permission has previously been granted.
The Department of Justice urges us to read 301 (f) as prohibiting a refusal to permit entry or inspection at any reasonable time. It argues that that construction is needed if the Act is to have real sanctions and if the benign purposes of the Act are to be realized. It points out that factory inspection has become the primary investigative device for enforcement of this law, that it is from factory inspections that about 80 % of the violations are discovered, that the small force of inspectors makes factory inspection, rather than random sampling …of finished goods, the only effective method of enforcing the Act.
All that the Department says may be true. But it does not enable us to make sense out of the statute. Nowhere does the Act say that a factory manager must allow entry and inspection at a reasonable hour. Section 704 makes entry and inspection conditioned on “making request and obtaining permission.” It is that entry and inspection which 301 (f) backs with a sanction. It would seem therefore on the face of the statute that the Act prohibits the refusal to permit inspection only if permission has been previously granted. Under that view the Act makes illegal the revocation of permission once given, not the failure to give permission. But that view would breed a host of problems. Would revocation of permission once given carry the criminal penalty no matter how long ago it was granted and no matter if it had no relation to the inspection demanded? Or must the permission granted and revoked relate to the demand for inspection on which the prosecution is based? Those uncertainties make that construction pregnant with danger for the regulated business.
The alternative construction pressed on us is equally treacherous because it gives conflicting commands. It makes inspection dependent on consent and makes refusal to allow inspection a crime. However we read 301 (f) we think it is not fair warning… to the factory manager that if he fails to give consent, he is a criminal. The vice of vagueness in criminal statutes is the treachery they conceal either in determining what persons are included or what acts are prohibited. Words which are vague and fluid…may be as much of a trap for the innocent as the ancient laws of Caligula. We cannot sanction taking a man by the heels for refusing to grant the permission which this Act on its face apparently gave him the right to withhold. That would be making an act criminal without fair and effective notice…
This Supreme Court opinion led Congress to pass the Factory Inspection Amendment in 1953. The Factory Inspection Amendment remains a critical provision some 60 years later. The Amendment introduced several new concepts. First, it removed the consent requirement from the FDA ’s inspection authority . Instead, it required the FDA to present credentials and a written notice of inspection. This written notice is known as Form 482 . The procedure created, and still used today, allows the FDA entry to any facility during a reasonable time simply by showing their credentials, typically an FDA badge, and the Form 482 . The criminal penalties were retained under the Amendment. It remains a criminal penalty to refuse any FDA agent making a request for entry under a Form 482. Second, the Amendment added a report of significant violations observed during the inspection. This is known as Form 483 “Inspectional Observations .”
The Form 483 stands as the most visible and widely recognized aspect of FDA investigations. It states the investigator’s opinion on any significant violations found in the facility during their visit. The Form 483 is reviewed by a compliance officer who makes a determination whether further enforcement actions, like a warning letter, are warranted. Prior to the Amendment following a facility inspection , owners were given no summary of violations which could foreshadow potential enforcement actions. This not only blindsided facilities when enforcement actions were taken many months later, but also risked misbranded or adulterated products leaving the facility.
Investigator or Inspector?
The terms are often used interchangeably when referring to the US FDA agent conducting a civil investigation . In either case, it will refer to the field personnel conducting the facility inspection . Within the FDA, however, there is a meaningful distinction between an “investigator” and an “inspector.” The FDA assigns an inspector to inspect “technologically noncomplex” facilities like bakeries and candy manufacturers and to collect samples or investigate routine consumer complaints (DeBell & Chesney).8 An investigator can complete those same tasks, but carries more training, a technical background, or advanced degree, which allows them to inspect more complex facilities or complaints like dietary supplement manufacturing. A facility may be visited by either type depending not only on the facility, but also the overall availability of personnel in the field office.
2.3.2 Where Capitalize Find the FDA
Unlike FSIS, the FDA is not in the facility on a daily basis. This raises the question, where do we find the FDA? The answer depends on the circumstances. Typically regulatory matters can be divided into two camps. The first camp, commonly called “premarket,” relates to the approval process for new ingredients and additives for example. It is important to note straightaway the FDA adopts the stance that it is not a consultative agency. Premarket submissions will be a discussion in a later chapter (see, Chap. 6). For now, be cognizant of the limitation to approach the FDA freely with questions and/or advice. The second camp, referred to as “postmarket surveillance,” encompasses all of the enforcement activities conducted by the agency. This includes inspections at foreign and domestic facilities, inspections of imports, and criminal matters.
The locus of premarket submissions is CFSAN. CFSAN is one of the six product-oriented centers that comprise the FDA . The other five are: CVM , Center for Drug Evaluation and Research (CDER) , Center for Biologics Evaluation and Research (CBER) , Center for Device and Radiological Health (CDRH) , and Center for Tobacco Products (CBT) . This organization intuitively informs industry and consumers the personnel within the agency focused on their regulated product. The bulk of interactions with CFSAN occur at the FDA’s campus in College Park , Maryland. CFSAN also operates research facilities in Laurel, Maryland, Bedford, IL, and in Dauphin Island, Alabama, but it is uncommon to interact with those facilities (About CFSAN). 9
The FDA relies on its Office of Regulatory Affairs (ORA) to manage postmarket surveillance activities. The ORA operates all FDA field activities, including facility inspections, foreign and domestic, as wells reviews of imported products. It works for all six centers, not just CFSAN. Thus, it conducts investigations of drug , medical device, cosmetic, biologics , veterinary products , and food, beverage , and dietary supplements . The result of limited budgets and personnel can lead to an investigator conducting inspections in five or six areas. It is not uncommon for such an overlap to lead to errors. The FDA organizes all of its functions, including the ORA, into regions (see Fig. 2.4). The five regions are further divided into 16 districts . Within each district there is both a district office and several field offices . Overall, there are approximately 1900 FDA offices throughout the US.
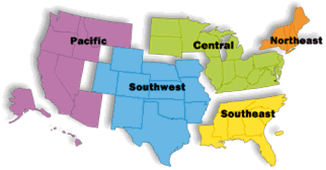
Fig. 2.4
US map broken into the five FDA regions
2.3.3 FDA Criminal Division
In addition to routine civil investigations the FDA also conducts criminal investigations. This function is conducted by the Office of Criminal Investigations (OCI) . Its scope of authority includes all FDA-regulated products, not just food, beverages, and dietary supplements . Criminal investigations are uncommon with civil enforcement actions occurring the most frequently. OCI concentrates its resources to four primary areas:
1.
Unapproved, counterfeit, and substandard medical devices and drugs;
2.
Escalation of civil enforcement actions where “the normal regulatory process has been unable to remedy the problem…”;
3.
Violations that post a significant risk to public health and the only remedy is criminal enforcement; and
4.
Impeding the FDA from properly performing its regulatory functions, such as providing “false statements to the FDA during the regulatory process and obstruction of justice” (OCI Mission) .10
In terms of food law and regulation OCI becomes involved when repeated civil enforcement actions fail to correct a violation. For example, following repeated investigations and warning letters from ORA the case will be transferred to OCI. Sans any significant risk to public health, this process usually follows several warning letters over a period of three or more years. OCI will then conduct its own investigations, including utilizing under-cover agents. Criminal actions will be discussed in detail in Chapter 7.
2.3.4 The Role of Form 483
The Factory Inspection Amendment gave birth to Form 483 . The same Amendment which gave the FDA authority to inspect and the written notice of inspection known as Form 482 also required Form 483. Section 374(b) states:
Upon completion of any such inspection … and prior to leaving the premises, the officer or employee making the inspection shall give to the owner, operator, or agent in charge a report in writing setting forth any conditions or practices observed by him which, in his judgment, indicate that any food, drug, device, tobacco product, or cosmetic in such establishment (1) consists in whole or in part of any filthy, putrid, or decomposed substance, or (2) has been prepared, packed, or held under insanitary conditions whereby it may have become contaminated with filth, or whereby it may have been rendered injurious to health. A copy of such report shall be sent promptly to the Secretary.
Form 483 at its simplest is a notice of potential violations found during an inspection. Form 483 is issued at the conclusion of a facility inspection by the inspector conducting the audit. It contains their opinion on significant violations , but is not reviewed by a compliance office, district director or any other FDA officer prior to being issued. Section 374(b) requires the reporting of adulteration observations , but the more common Good Manufacturing Practices (GMPs) violations are listed by FDA policy. The IOM provides further guidance to the investigator on what observations to cite. In Section 5.2.3 the IOM directs investigators to adhere to two general principles.
IOM 5.2.3 General Principles of Reporting Observations
Observations which are listed should be significant and correlate to regulated products or processes being inspected.
Observations of questionable significance should not be listed on the FDA-483, but will be discussed with the firm’s management so that they understand how uncorrected problems could become a violation. This discussion will be detailed in the EIR.
There are some observations considered violations that are not reported in Form 483 . These violations could serve as the basis for a warning letter or other enforcement action. The policy holds that the violations should not be reported because they require further review before citing. The IOM provides a list of these non-reportable violations in Section 5.2.3.3.
IOM 5.2.3.3 Non-Reportable Observations
1.
Label and labeling content with some exceptions.
2.
Promotional materials.
3.
The classification of a cosmetic or device as a drug.
4.
The classification of a drug as a new drug.
5.
Non-conformance with the New Drug Regulations, 21 CFR 312.1 (New Drugs for Investigational Use in Human Beings: Exemptions from Section 505(a)) unless instructed by the particular program or assignment.
6.
The lack of registration required by Section 415 and 510 of the FD&C Act. The lack of registration per 21 CFR 1271 Subpart B Procedures for Registration and Listing, promulgated under Section 361 of the PHS Act.
7.
Patient names, donor names, etc. If such identification is necessary, use initials, code numbers, record numbers, etc.
8.
Corrective actions. Specific actions taken by the firm in response to observations noted on the FDA 483 or during the inspection are not listed on the FDA 483, but are reported in the EIR . Except as described in IOM 5.2.3.4.
9.
The use of an unsafe food additive or color additive in a food product.
Form 483 is used by an investigator to write an Establishment Inspection Report (EIR) . Together the EIR and Form 483 inform a compliance officer or district director of potential violations. The EIR will experience substantial review to determine whether a warning letter or other enforcement action is required. As a matter of policy a company may submit a response to the FDA within 15 days of receiving Form 483. A response is not required, but if not given the EIR and Form 483 will only provide the FDA compliance officer one side of the story. An appropriate response can avoid or mitigate further enforcement action while a fumbled response can exacerbate or hasten additional enforcement activity.
As the formal name of Form 483 suggests its application is limited to facility inspections. It will be used for any facility inspection , foreign or domestic, for-cause or surveillance, but not in any other situation. It is not utilized, for example, in the case of import refusals or detentions. Imports will involve a separate set of forms and notices. An import refusal may only trigger an inspection, which could lead to a Form 483. This attenuated example is as close as a Form 483 comes to imports.
The primary purpose of adding Form 483 to the Factory Inspection Amendment was notification of potential errors. Prior to the Amendment an inspection would occur with the FDA leaving no indication of the inspection findings. This left formal enforcement as the only means for facilities to engage in a discussion about minor violations that could be corrected quickly, potential errors made by the investigator, or adopt a proactive stance to get ahead of major violations. Formal enforcement actions are time and cost intensive along with being incredibly public. Formal enforcement actions can also be slow to initiate and create a lengthy gap between when the violation was observed and when corrective actions are sought. In short Form 483 stepped in to fill an important policy shortcoming
How to Respond to a Form 483
As a prewarning of enforcement action Form 483 offers facilities an opportunity to avoid more burdensome enforcement actions. Here are some tips in responding to a Form 483:
Avoid the Three Stooges response: Rather than firing off a response before reviewing the regulations , observations , and corrective actions be calm, clear, cogent addressing each observation separately;
Avoid the Sue Sylvester anger-first response: An inspection can feel deeply personal, like an attack, especially for small and medium-sized facilities. Drop the anger-first response. Verbally assaulting the FDA, the investigator, or jumping to broad illegalities will not resolve Form 483 and likely serve to make the enforcement matter worse.
Avoid the Urkel “Did I Do That?” Response: Some facility owners adopt the idea that playing dumb will garner pity from the agency and make the whole matter disappear. This typically involves a response with a series of “Did I Do That?” and statements disclaiming any knowledge of the regulations . Instead of pity the FDA quickly becomes concerned by a facility that appears to lack control of its operations. Show command of the issues, the facility, and the potential crisis at hand. Confidence rather than ignorance will grease the wheels in quickly resolving a Form 483. .
In many instances Form 483 serves as the precursor to additional FDA enforcement action. Although not required by the regulations , it often works in tandem with a warning letter. The common escalation of enforcement actions, for facility inspections, at least begins with Form 483 followed by a warning letter and potentially additional formal enforcement actions (see Fig. 2.5). A Form 483 will escalate to a warning letter where the observations are too serious to be resolved at the Form 483 stage or the response indicates further FDA involvement is required.

Fig. 2.5
Inspection progressing to a Form 483, warning letter and its relationship with the FDA criminal division
2.3.5 Warning Letters as Enforcement Instruments
Warning letters are unofficially the first enforcement tool used by the FDA . This is not necessarily how the FDA views warning letters. Under FDA policy warning letters are merely considered “informal enforcement actions ” simply intended to provide a recommendation on the steps need to achieve compliance. These informal recommendations elicit real changes that impact facilities in much the same way as formal enforcement actions. Yet, for the agency the warning letters simply function as “prior notice.”
Prior notice serves both as notice and an ultimatum. The Regulatory Procedures Manual (RPM) describes prior notice in Chapter 10. There the agency states the practicality of prior notice. Practical because it compels or induces voluntary compliance and acts a place-holder or evidence of violative behavior all without the time or cost of formal enforcement actions. Prior notice is not a statutory requirement but an administrative agency’s own policy. The FDA can and does take formal enforcement action without warning letters.
Prior notice is not appropriate in all circumstances. In some cases immediate formal enforcement action must be taken. In others an accumulation of warning letters or Form 483s serve as enough notice. Outside of the mundane ordinary cases prior notice does not work well. Where there is evidence of criminal or intentional violations, for example, the agency will take formal action first. Likewise where there is a reasonable and real threat to the public, the agency pounces rather than provide prior notice. The RPM in subchapter 4-1-1 provides other examples on when a warning letter may be inappropriate.