Adulteration
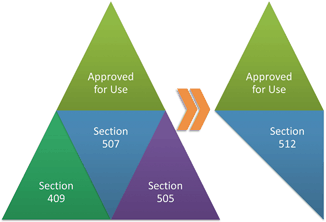
Fig. 3.1
Changes between 1906 and 1938 Act
When drafting the 1938 Act Congress wanted to provide the FDA greater control over certain added substances. As will be discussed in detail in Section 4 the “may render injurious” standard is less rigorous and permits a wide range of additives into food compared to the “ordinarily injurious” test. Congress enacted a type of license scheme in Section 406 of the 1938 Act. Section 406 provides the FDA to establish tolerances for added “poisonous or deleterious ” substances only when the substance is “required” or “cannot be avoided” (FD&C) .3 Thus, for substances that are useful or difficult to eliminate, the FDA can regulate tolerances to ensure the additives remain safe.
The food adulteration definition was originally codified in Section 402 . All subsequent amendments follow in sequence. For instance the Pesticides Residue Amendment was codified as Section 408 . The adulteration section is currently codified as Section 342 with all Amendments codified in sequential order beginning with Section 346 . When searching for definitions and case law be aware of the original codification, which is still often used as a reference, and the current location of the adulteration regulations.
Overview of the Amendments
The 1938 Act provides a triad of controls that functioned in the market for nearly 20 years. It lists adulteration as a prohibited act in Section 331. A substance under the 1938 Act was adulterated if an added substance failed the “may render injurious” test, if a non-added substance fumbled the “ordinarily injurious” standard, or in limited cases where added substances regulated under a tolerance exceeded the maximum threshold (see Fig. 3.2). This framework sufficed from 1938 until the first Amendment was passed in 1954.
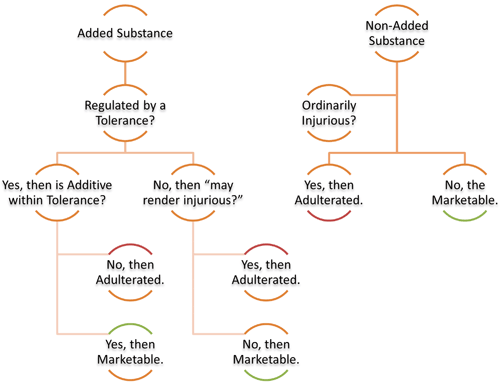
Fig. 3.2
Initial decision tree to determine adulteration
Beginning in 1954 Congress began considering and passing a series of adulteration amendments, a process that would last over the course of six years. It began with the Pesticides Residues Amendment in 1954. The Pesticides Amendment changed the criteria for finding raw agricultural commodities (RACs) adulterated. Originally enacted as Section 408 it is currently codified as Section 346a . The Pesticides Amendment specifically amended Section 406 , which authorized the agency to set tolerances . The Amendment deemed a RAC adulterated if it contains a pesticide residue in excess of the tolerances established in the Amendment. The Amendment provides a complex system for determining the tolerances of various pesticides.
Two years later Congress passed the Food Additives Amendment . The Amendment was originally codified as Section 409 but is currently found in Section 348 . The Amendment applies to a wide range of food substances, including food packaging materials . It is a complex system that will be addressed as a separate topic in Chapter 6. For our purposes in Chapter 3 it suffices to say any food additive not approved under the food additive licensure scheme or exceeding the limits of its approval is adulterated.
The Food Additives Amendment also added a new definition to the Act. Prior to 1958 the Act did not need and did not define the term “food additive.” Adulteration pertained only to foods as defined in the Act. The food additive definition is found in Section 201(s). The definition sweeps in both direct and indirect additives . Food packaging is an example of an indirect food additive . The main criteria for determining whether the added substance qualifies as a food additive under the Act is whether it exercises a technical effect on the food it is added to.
Food Additive Definition in Section 201(s)
“food additive” means any substance the intended use of which results or may reasonably be expected to result, directly or indirectly, in its becoming a component or otherwise affecting the characteristics of any food (including any substance intended for use in producing, manufacturing, packing, processing, preparing, treating, packaging, transporting, or holding food; and including any source of radiation intended for any such use)…
The final amendment came in 1960 when Congress passed the Color Additive Amendment . The Color Additive Amendment split apart color additives from the 1958 Amendment. The result was a stricter system of approval and use of color additives. Whereas food additives, those that are not used primarily to impart color, can find approval through a series of exemptions (see Chap. 6), color additives are not subject to any safe harbors. Color additives must be approved and listed by the FDA . Otherwise the color additive will be deemed adulterated. Chapter 6 will discuss color additive approval in more detail.
Color Additive Amendments broke from the sequential numbering model of prior Amendments. The Color Additive Amendment was originally codified as Section 721 far from Section 409 . This was in part because color additive regulations applied to all FDA regulated products, not just foods bearing color additives . The 1960 Amendment is now codified as Section 379e .
Although not a separate Amendment the Delaney Clauses deserve a separate discussion. The Delaney Clauses are named after Congressman James “Jim” Delaney (see Fig. 3.3) . The first Delaney Clause is found in the Food Additives Amendment . It precludes the FDA from approving any additive that may be found to induce cancer in humans or experimental animals. The Delaney Clause is an addition to the Food Additives Amendment and only applies to substances governed by Section 409 . The Delaney Clause was also added to the Color Additive Amendment.

Fig. 3.3
Image of Congressman Jim Delaney
The final and most recent adulteration Amendment to the 1938 Act came 30 years later when Congress passed the Animal Drug Amendment . Following the passage of the Food Additives Amendment, the FDA regulated drugs administered to food -prod ucing animals under Section 409 and the appropriate drug approval provisions. Section 409 applies to drugs administered directly to animals that could “reasonably be expected” to leave residues in human food. The 1968 Amendment simplified the process. Under the Amendment a single approval system for animal drugs was implemented. The Amendment also prohibited the sale of drugs likely to leave residues and the sale of food containing residues absent FDA approval .
The new system of original definitions and Amendments created a complex system (see Fig. 3.4). In the majority of cases it can be challenging to define exactly why a product is adulterated. Even the seemingly straightforward example layers of exemptions and statutory criteria require verification and analysis. It is important to understand the web of regulations both for enforcement purposes and when developing a new product.
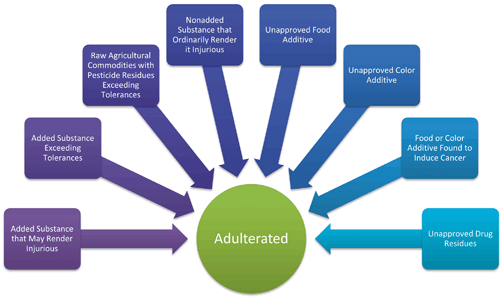
Fig. 3.4
Adulteration framework
The USDA and Adulteration and Food Additives
Notably absent from the discussion above is any mention of the USDA and FSIS . Each of the enabling acts controlled by FSIS contains a definition of adulteration . FSIS remains in primary control for inspecting and enforcing adulterated meat, poultry, and eggs. The definitions of adulteration are identical to the definitions used by the FDA .
Poultry Products Inspection Act Section 453(g)(1)
Poultry product is adulterated “ … if it bears or contains any poisonous or deleterious substance which may render it injurious to health ; but in case the substance is not an added substance, such article shall not be considered adulterated under this clause if the quantity of such substance in or on such article does not ordinarily render it injurious to health…”
Federal Meat Inspection Act Section 601(m)(1)
Meat or meat food products are adulterated “ … if it bears or contains any poisonous or deleterious substance which may render it injurious to health ; but in case the substance is not an added substance, such article shall not be considered adulterated under this clause if the quantity of such substance in or on such article does not ordinarily render it injurious to health…”
Egg Products Inspection Act Section 1033(a)(1)
Egg or egg products are adulterated “ … if it bears or contains any poisonous or deleterious substance which may render it injurious to health ; but in case the substance is not an added substance, such article shall not be considered adulterated under this clause if the quantity of such substance in or on such article does not ordinarily render it injurious to health…”
The FDA remains the primary agency for regulating the use of food additives even in meat and poultry. The FDA and FSIS share responsibility for the safety of food additives used in meat, poultry, and egg products. The FDA, however, sets the minimum safety regulations which FSIS must enforce. Under the Food Additives Amendment all proposed additives are first evaluated by the FDA. The FDA will determine whether or not the additives may be used. A secondary review may be conducted by the Risk, Innovations, and Management division of FSIS.
FSIS can only elect to apply stricter food additive standards than those adopted by the FDA . As experts in meat, poultry, and egg products, FSIS believes it can make better determinations about the technical effects of additives proposed for FSIS regulated products. Thus, it may elect to set higher standards than what the FDA sets for a food additive. When a higher standard is adopted an additive must be approved first by the FDA, then by FSIS. FSIS may never set a lower standard than the one set by the FDA. FSIS provides the example of sorbic acid on its website (FSIS Additives).4 The FDA approved sorbic acid as a food additive. But in order to use ’this product in meat, any applicant seeking to use it would need approval from FSIS who adopted a more stringent sorbic acid standard. FSIS set a higher standard for sorbic acid in meat out of concern that the additive could “mask spoilage caused by organisms that cause foodborne illness” (FSIS Additives).5
3.2 Types of Adulteration
The definition of adulteration provided in Section 1 above was intentionally incomplete. It addressed only the poisonous and de leterious standard for added or naturally occurring (non-added) substances. It omitted the remaining provisions addressing other means by which food becomes adulterated. In Subsections 3 and 4 the Act deems adulterated facility conditions that could indirectly contribute to adulteration. Subsection (b) regulates the concept of economic adulteration. A final type of adulteration considered by the agency is intentional adulteration or contamination. This can take several forms, including acts of terrorism , disgruntled employees, consumers, or competitors. It captures an act done with knowledge of the potential injury or harm to others. As will be discussed below the FDA ’s policy is for facilities to adopt food defense or food security measures to protect against intentional adulteration. Economic adulteration is often swept into this concept since it is clearly an intentional act, but for purposes of this text will be treated separately.
21 U.S.C. 342(a)(3)-(5);(b)
…or (3) if it consists in whole or in part of any filthy, putrid, or decomposed substance, or if it is otherwise unfit for food ; or (4) if it has been prepared, packed, or held under insanitary conditions whereby it may have become contaminated with filth , or whereby it may have been rendered injurious to health ; or (5) if it is, in whole or in part, the product of a diseased animal or of an animal which has died otherwise than by slaughter…
(b) Absence, substitution, or addition of constituents
(1) If any valuable constituent has been in whole or in part omitted or abstracted therefrom; or (2) if any substance has been substituted wholly or in part therefor; or (3) if damage or inferiority has been concealed in any manner; or (4) if any substance has been added thereto or mixed or packed therewith so as to increase its bulk or weight, or reduce its quality or strength, or make it appear better or of greater value than it is.
The 1938 Act encompassed a comprehensive view of adulteration . Its provisions regulated scenarios where substances were added, naturally occurring , and posed a risk of contamination during production. The Act ensured the public’s safety from harmful products and from products whose only harm was economic. The sections below will look closer at the new concepts of intentional, indirect, and economic adulteration.
3.2.1 Economic Adulteration
Economic adulteration involves the selling of a product containing inferior ingredients but marketed as containing superior ingredients. It is an attempt to confuse the consumer and profit from the fraud . This type of pass-off offense finds ancient roots in China, Greece, and Rome. Examples in the early 1900s when the Pure Food and Drug Act was passed include milk diluted with water or maple syrup diluted with cane sugar. In most cases there is no risk of harm.
Law of Moses
The fat of an animal found dead or torn by wild animals may be used for any other purpose, but you must not eat it. (Leviticus, 7:24.)
Ancient Chinese Literature and Legal Code
“The Supervisor of Markets had agents whose duty it was to prohibit the making of spurious products, and the defrauding of purchasers.”
When dried or fresh meats cause men to become ill, all the leftover portions should be speedily burned. The violator will be flogged 90 strokes. He who deliberately gives or sells it to another will be banished for a year, and if the person to whom it has been given or sold dies, the offender will be hanged…
Roman—Pliny the Great
“…the dealers have set up regular factories where they give a dark hue to their wine by means of smoke, and, I regret to say, employ noxious herbs, in as much as a dealer actually used aloe for adulterating the flavor and color of his wine.”
Pliny also provides examples of wine diluted with water and flour mixed with chalk to increase its whiteness and shortness.
The lack of harm makes economic adulteration unique. In general a food is deemed adulterated when it contains a “poisonous or deleterious ” substance that could pose a health risk. In many cases economically adulterated products lack this risk. There are early examples, such as ink dyes used as coloring agents, which were injurious to both body and purse. Arguably such a case today would be treated as intentionally adding an adulterant rather than economic adulteration. In fact, it is a struggle to find cases where the FDA uses its limited resources to pursue economic adulteration. This is because the resources are directed to the products posing a risk of physical injury, which economic adulteration lacks.
Sole Economic Harm Not Classified as Misbranding
Economic adulteration may be a misnomer. Typically when the Act enforces provisions related to economic harm, deception, or fraud where there is no physical risk, it does so as misbrandings . Misbrandings will be discussed in the next chapter, but can generally be described as labeling offenses. The distinction between misbranding and adulteration lies in the enforcement tools the agency can use.
Adulteration allows the agency to move quickly with little proof of adulteration . The bulk of adulteration refers to areas of potential injury, which requires immediateand swift action. Thus, the FDA can make multiple seizures or administrative detention of adulterated foods . Misbranded items in many cases lack the immediacy of adulteration. Some cases like labels failing to declare an allergen may be an exception. When the concept is limited to fraudulent claims, imposing only economic harm it is much harder for the FDA to act.
Economic adulteration shares the characteristics of misbranding but carries the enforcement triggers of adulteration. It is a hybrid creature born of the circumstances of the 1906 Act. Economic adulteration bears little, if any, difference between other fraudulent statements made on a label. It relates to ingredients, whereas other claims may relate to health benefits or product features. Yet for no reason other than historical happenstance the use of inferior ingredients passed off as superior ones is treated as adulteration. Only when considering the history of the 1906 Act does this make sense.
The 1906 Act arouse out of a tumultuous period of food production. This text in previous chapters described the findings of Upton Sinclair . It was not only in meat packing plants that deplorable conditions and acts were found. It was fairly pervasive throughout the food industry. It is likely that this lingering fear and revulsion of abuse of trust in food production is what continues to keep these acts classified as adulteration .
Concurrent Jurisdiction—FDA and Federal Trade Commission
Economic adulteration is the first area of law where we encounter concurrent jurisdiction with a federal agency that is unaffiliated with food regulation . In 1914 Congress provided the Federal Trade Commission (FTC ) to enforce deceptive commercial practices. This came in the form of the Wheeler Lea Act. The FTC interprets its enabling acts to provide it authority to stop the sale of food products that deceive the public or provide the seller an unfair advantage in the market.
The FDA exercises primary control over economic adulteration . As will be seen with misbranding, the FTC largely defers to the FDA on economic adulteration. The agencies operate under a working agreement which outlines the boundaries of authority (FDA-FTC MOU).7 Under the working agreement the two agencies will either act concurrently or through case referrals. The FTC claims jurisdiction over advertising and marketing and the FDA ischarged with regulating the labeling. Labeling leaves the FDA in control of the standards on what constitutes economic adulteration. The reason being the front of the label will declare the product’s identity, such as blueberry muffins, and the ingredient panel will declare ingredients , like blueberry bits (see Modern Examples on p.78). This is the only area of adulteration where the FTC can exercise authority. In every other place the FTC can act in relation to marketing and claims. This again raises the question why economic adulteration is not a misbranding offense. The two agencies are moving towards cooperation rather than competition, as will be discussed in Chapter 4 even issuing joint warning letters.
Problematic 1906 Act
The 1906 Act attempted to regulate economic adulteration . The Act began with a general definition of adulteration. Language like “substitutions,” however, could be broadly interpreted to preclude any modern developments in food production. Seemingly aware of the impediment Section 7 the drafters added two important exemptions.
Section 7 of the Pure Food and Drug Act (1906)
First. If any substance has been mixed and packed with it so as to reduce or lower or injuriously affect its quality or strength.
Second. If any substance has been substituted wholly or in part for the article.
Third. If any valuable constituent of the article has been wholly or in part abstracted.
Fourth. If it be mixed, colored, powdered, coated, or stained in a manner whereby damage or inferiority is concealed…
The exemptions were necessary to make the economic definition work but were also prone to abuse. Section 8 on misbranding provided two labeling requirements which, if followed, allowed modern processed foods. The first exemption is known as the “distinctive name” exemption . The most infamous example is “Bred Spread” which is neither peanut butter nor jam, but its own distinctive name (see Fig. 3.5). Another early example was “Grape-Smack” which was imitation grape juice. Section 8 allowed a fabricated food on the market if it was labeled with a distinctive name. A second provision allowed compounds, imitations, or blends if proper labeled. Thus, rather than “butter” a label could bear the statement “imitation butter.” Under the distinctive names and imitation exemptions honest manufacturers could develop and sell new innovative foods. It also worked to effectively gut the economic adulteration provisions.
What is Hummus?
Sabra Dipping Co., LLC submitted a citizens petition requesting a standard of identity for hummus. Currently a variety of products sell under the common name “hummus” without needing to adhere to a standard of identity. Sabra seeks to identify hummus as consisting of chickpeas and no less than 5 % hummus. The 11-page petition can be found using Docket FDA-2014-P-0259.

Fig. 3.5
Examples from the FDA of distinctive names
Enforcing the distinctive names exemption proved challenging. Manufacturers quickly learned to adopt distinctive names and claim compliance with Section 8. Grape-Smack, for example, escaped enforcement for economic adulteration . The imitation grape juice contained calcium acid phosphate and corn starch, but was labeled with the initials of a more expensive brand, “C.A.P.” (100 Barrels of Calcium Acid Phosphate).8 The courts were soon flooded with cases focused not on the inferior ingredients or alleged fraud , but on whether names likes “Macaroons” or “Maple Flavo” were distinctive names. This was an abstruse and bizarre consequence of the Section 8 distinctive names exemption.
The second exemption did not fare any better in court. The exemption was written broadly to provide safe harbor to any product labeled “compound,” “imitation,” or “blend.” Some courts interpreted this language plainly giving shelter to any product appropriately labeled. Other courts read Sections 7 and 8 together and required the ingredients of the compound to be listed. For instance one court found “Fruit Wild Cherry Compound” (Weeks)9 not economically adulterated because it was labeled “compound,” but a second court found “Compound Ess Grape” (Schider)10 economically adulterated despite complying with Section 8 because it listed a single ingredient imitation grape essence. Litigation was unpredictable and the ease of challenging the FDA left few cases resolved administratively.
Improvements in the 1938 Act
For over 30 years courts, industry,` and the FDA suffered under the 1906 economic adulteration fiasco. The 1938 Act directly addresses all of the concerns raised, enforcing the economic adulteration provisions between 1906 and 1938. It resolves all of the confusion and abuses of the 1906 Act. It began by striking the distinctive names and imitation exemption, then added new provisions to take their place.
The 1938 Act replaced distinctive names with standards of identity . The FDA gained new authority to created standards of identity for each food product following public notice and hearing. No longer would “Bred Spred” escape enforcement as economically adulterated. If it did not meet the standard of identity for peanut butter or jam then it was economically adulterated. It could continue to be its own non-standardized product, but it would need to label itself imitation and state its ingredients on the label. Thus a product either meets a standard of identity or must be labeled imitation and list all its ingredients. It could also petition the FDA under 21 CFR 130.5 to create a new standard of identity for its product. The petition is not unique to standards of identity but is called a citizens petition , which can be used for a variety of purposes.
The same definition of economic adulteration from the 1906 Act continues today. The 1938 Act did not change the overall definition of economic adulteration. It remains as broad and unworkable today as it was when enacted over a hundred years ago. Absent the exemptions to provide some boundaries defining economic adulteration, the provision is ambiguous. Many commentators describe economic adulteration as too broad to interpret plainly and too ambiguous to be interpret intelligently.
Modern Examples
Economic adulteration is not a relic of the past. It remains as vibrant and attractive cost-saving measure as it was in ancient Rome, Greece, or China. Many modern examples highlight the use and lack of enforcement over economic adulteration. For instance, blueberries are an expensive but highly marketable ingredient. Read your labels carefully. Does that package list real blueberries? In 2011 a consumer advocacy group found several products claiming blueberries, such as blueberry bagels, muffins, and cereals that did not contain actual blueberries. Many labels instead claimed “blueberry bits” which included sugar, food dye, and corn syrup. Unsuspecting consumers would buy the product believing it contained a superior ingredient, blueberries, when it actually contained an inferior ingredient, blueberry bits. This lead to several labeling lawsuits as discussed in Chapter 7. Blueberry bits are a classic example of economic adulteration and one that went unenforced by the FDA .
3.2.2 Indirect Adulteration—Filth and Insanitary Conditions
Adulteration by filth or insanitary conditions is made for television material. It involves exactly the worse we can imagine—facilities infested with mice or cockroaches, or food contaminated with objects, insects, or worse. This type of adulteration echoes The Jungle in finding the worst of food production.
FSIS and the FDA share the same definition for indirect adulteration . Indirect adulteration, it should be noted, is distinct from indirect food additives. Rather than discussing Section 409, food additives, here the provisions for indirect adulteration are found in Sections 402(a)(3) and (4). As with the all aspects of adulteration the FMI, PPI, EPI mirror the FD&C . Therefore both agencies are operating under the same concept of adulteration by filth and insanitary conditions.
The FDA organizes indirect adulteration under 402(a)(3) and (4) into three subcategories. When evaluating adulteration the FDA will look for health hazards , indicators of insanitation , and natural or unavoidable defects . The health hazard category includes physical , chemical , and microbiological hazards associated with extraneous materials. Insanitary conditions include criteria like visibly objectionable contaminants and evidence of infestations. The health hazard category encompasses criteria from the Hazard Analysis and Critical Control Point (HACCP ) system. All three categories are subject to control.
Defect Action Levels for Filth
Most filth is unavoidable. As unsettling as it may seem food will never be defect-free. Insect parts for example are an accepted element of harvesting everything from grapes to cocoa. Similar to the tolerances for added substances, the FDA sets “action levels ” for unavoidable poisonous or deleterious substances. Tolerances and action levels are similar in that both set a maximum level for a substance that could pose harm. Tolerances, however, as intentionally added substances are subject to a formal rule. The FDA will provide public notice and hold hearings before setting a tolerance. The tolerance is then giving the full force of law as if it were part of the Act. Action levels are informal judgment on the quantities of a particular contaminant that will not pose a health risk. Action levels, therefore, represent a promise by the FDA not to enforce any level of contamination from unavoidable sources but only commit to formal enforcement when a threshold or action level is crossed.
Action levels only apply to the unavoidable or natural defect category. The FDA publishes all of the action levels in a guidance document . If the contaminant is not subject to an action level, the enforcement discretion will not apply. This includes blending a food product containing a substance in excess of an action level with another food product in an attempt to dilute the contaminant (FDA Guidance).11 Action levels can also be found in Parts 109 and 509 of the CFR.
Examples of FDA Action Levels (FDA Guidance)12
LEAD | ||
|---|---|---|
Commodity | Action level (µg/ml leaching solution) | Reference |
Ceramicware | ||
Flatware (average of 6 units) | 3.0 | CPG 545.450 |
Small hollowware (other than cups and mugs) (any 1 of 6 units) | 2.0 | CPG 545.450 |
Large hollowware (other than pitchers) (any 1 of 6 units) | 1.0 | CPG 545.450 |
Cups and mugs (any 1 of 6 units) | 0.5 | CPG 545.450 |
Pitchers (any 1 of 6 units) | 0.5 | CPG 545.450 |
Silver-plated hollowware | ||
Product intended for use by adults (average of 6 units) | 7 | CPG 545.450 |
Product intended for use by infants and children (any 1 of 6 units) | 0.5 | CPG 545.450 |
MERCURY | ||
|---|---|---|
Commodity | Action level | Reference |
Fish, shellfish, crustaceans, other aquatic animals (fresh, frozen or processed) | 1 ppm methyl mercury in edible portion | CPG 540.600 |
Wheat (pink kernels only) | 1 ppm on pink kernels and an average of 10 or more pink kernels/500 g | CPG 578.40 |
Good Manufacturing Practices (GMPs, or cGMPs) and Insanitary Conditions
Good Manufacturing Practices are the primary control to maintain minimum sanitary conditions in a facility. The Good Manufacturing Practices are published in 21 CFR 110 for food and 111 for dietary supplements . The GMPs outline all of the sanitation controls and methods for equipment, grounds, shipping, and facility. An entire GMP is dedicated to pest control, the most prevalent citation for insanitary conditions .
GMPs are important for many reasons. The obvious value in GMPs is providing a uniform safety net to ensure all food products are produced, stored, and shipped in a way that protects the integrity and wholesomeness of the food. GMPs are also important because the procedures serve as invisible link in the bond of trust between consumer and producer. A consumer cannot visit every food facility that makes the product in their cupboard. The global economy is about production occurring away from our homes and communities by a number of actors we trust but cannot see. Neither can consumers see the effects of that facility—the rats near finished product, the cockroaches near production lines, the mold on the floor or ceiling, and so on. GMPs when followed provide the public confidence that the facility their food originated at is a clean facility. This is often why GMP violations are prevalent in Form 483 .
FSMA and the Hazard Analysis Risk-Based Controls Rule
Identifying and controlling hazards ensures food is safe. Hazards can include sanitation practices, but also a variety of issues unique to particular product or class of products. Soft cheeses, for example, are prone to contamination with Listeria or seafood, which requires refrigeration at specific temperatures. Failing to control facility-wide and product-specific standards allows potential hazards into a food product, including chemicals, physical hazards, and microbiological contamination.
FSMA fundamentally changes how hazards are controlled. Prior to FSMA, facilities could voluntarily adopt a HACCP plan, which aims at identifying and controlling hazards. Otherwise there was a risk of adulteration , but there was no enforcement or mechanism to ensure that hazards were controlled. FSMA changed the existing paradigm by introducing a new rule known as the Hazard Analysis Risk-Based Control Rule , or Preventative Controls rule . The Preventative Controls rule implements two changes. It revises the GMP regulations with minor tweaks and more importantly introduces a mandatory hazard control system.
The Preventative Controls rule essentially introduces a HACCP -esque system for qualifying food facilities. The rule varies from HACCP in key areas, but the overall aim is the same. The rule requires science and risk-based preventive controls as necessary to prevent hazard associated with a facility and its products. Not all facilities are subject to the rule with exemptions for small and very small facilities. There are also modified requirements for certain low-risk activities. A facility must identify only those hazards “reasonably likely” to occur. It is then given flexibility in what preventative controls are used, but the FDA will assess the adequacy of the control. Hazard identification and control will soon be a mandatory means for ensuring against indirect adulteration .
3.2.3 Intentional Adulteration
Intentional adulteration involves any number of acts taken with the purpose of causing harm. This is not the typical case of adulteration. Instead, it is the rare case that requires vigilance to protect against the widespread harm that could result from such an act. It can involve a person slipping away in a transport vehicle, tampering with food in the grocery store, or introducing an adulterant in a production facility.
Interagency Approach
Intentional adulteration typically involves acts beyond a food facility’s or the FDA ’s control. Acts of terrorism or tampering with products in a grocery store require an interagency approach. The FDA and USDA coordinate with a long list of agencies on food defense, including the Center for Disease Control and Prevention (CDC ), the Department of Homeland Security (DHS ), the Federal Bureau of Investigation (FBI ), the Environmental Protection Agency (EPA ), the Department of Defense , the Department of Energy , the Department of Commerce , and the Department of the Interior . The coordination also envelops state, local, tribal, territorial, and private sector partners to develop food safety plans. The FDA for its part completed vulnerability assessments of a variety of products and processes within the food and agriculture sector along with developing several guidance documents for industry.
Guidance Documents
Prior to FSMA the main tool for the FDA to address intentional adulteration was through Guidance Documents. The FDA issued five Guidance Documents and launched a new ALERT system to assist industry in protecting the food supply against intentional adulteration . All five Guidance Documents were issued in 2003. Each Guidance Document addressed food security preventive measure for specific types of facilities. It includes guidance for producer, processors, and transporters, importers, milk industry, retail food stores, and food service establishments, and the cosmetic industry. The Guidance Documents outline best practices in food defense and preventative measures to reduce the risk of intentional adulteration. The Guidance Documents were updated most recently in 2007. The FDA also developed a number of training programs and resources for State and industry partners.
FSMA and the Rule for Focused Mitigation Strategies to Protect Food Against Intentional Adulteration
FSMA introduced a new rule to mandate preventative controls aimed at mitigating the risk of intentional adulteration . The rule requires facilities covered by the rule to develop Food Defense Plans . The FDA identified four key activities most vulnerable to intentional adulteration .
Four Activities Identified in the Proposed Rule
1.
bulk liquid receiving and loading;
2.
liquid storage and handling;
3.
secondary ingredient handling (the step where ingredients other than the primary ingredient of the food are handled before being combined with the primary ingredient); and
4.
mixing and similar activities.